Difference between revisions of "Dominic Röhm"
| Line 34: | Line 34: | ||
== Publications == | == Publications == | ||
<bibentry> | <bibentry> | ||
| + | rouetleduc14a | ||
rohm12a | rohm12a | ||
roehm13a | roehm13a | ||
Revision as of 13:04, 20 March 2014

PhD student
| Office: | 1.077 |
|---|---|
| Phone: | +49 711 685-67705 |
| Fax: | +49 711 685-63658 |
| Email: | Dominic.Roehm _at_ icp.uni-stuttgart.de |
| Address: | Dominic Röhm Institute for Computational Physics Universität Stuttgart Allmandring 3 70569 Stuttgart Germany |
Research interests
Investigation of the heterogeneous nucleation, i.e. the early stage of crystal growth, in a colloidal model system near a wall by Molecular Dynamics computer simulations. My focus is on the influence of the hydrodynamic interaction, which is often neglected, since nucleation is considered as a quasi-static process.

However, recent experiments have shown, that the kind of thermalization of the sample has a drastic influence on the nucleation rate. The rate decreases by an order of magnitude, if the sample is molten by a turbulent flow rather than a laminar flow. In our simulations, hydrodynamic interactions are incorporated by coupling the particles to a lattice fluid representing the solvent.
Since the computation of the hydrodynamic interaction is still orders of magnitude more expensive than classical interactions, our MD simulation software ESPResSo employs GPUs for the calculation of the lattice fluid, which was realized during my diploma thesis. By this, we can investigate systems, that previously required a small computer cluster, using a single GPU. By using a cluster equipped with GPUs, we want to use this speedup to systematically investigate the influence of hydrodynamics on the heterogeneous nucleation. Another field of interest are heterogeneous multi-scale methods to model shock-wave propagation in metals. The multi-scale simulation is based on a non-oscillatory high-resolution scheme for two-dimensional hyperbolic conservation laws proposed by Jiang and Tadmor. I applied this general framework to a set of differential equations for elastodynamics in order to model materials under extreme conditions. Me and my collaborators computed the evolution of a mechanical wave in a perfect copper crystal on the macro-scale by evaluating stress and energy fluxes on the micro-scale. A finite-volume method was used as the macro-scale solver, which launches for every volume element a light-weighted MD simulation (called CoMD) to incorporate details from the micro scale. Since the execution of an MD simulation is rather costly, we reduced the number of actual MD simulations through the use of an adaptive sampling scheme. Our adaptive scheme utilizes a key-value database for ordinary Kriging and a gradient analysis to reduce the number of finer-scale response functions. Additionally, a mapping scheme has been implemented to handle duplicated calls of the MD simulation efficiently.

Kriging, also known as Best Linear Unbiased Prediction (BLUP), estimates an unknown value at a certain location by using weighted averages of the neighboring points. It also provides an error estimate, which we use to decide whether a value is accepted or if we have to evaluate the finer-scale response function with the help of CoMD. So far I focus on how the accuracy of the physical values is affected by several thresholds in our adaptive scheme and their connection to the overall performance. It seems that the enhanced adaptive scheme is sufficiently robust and efficient to allow for the future inclusion of details present in real materials, e.g. interstitials, vacancies, and phase boundaries.
Diploma Thesis
Lattice-Boltzmann-Simulations on GPUs
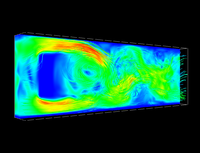
In coarse-grained Molecular dynamics (MD) simulations of large macromolecules, the number of solvent molecules is normally so large that most of the computation time is spent on the solvent. For this reason one is interested in replacing the solvent by a lattice fluid using the Lattice-Boltzmann (LB) method. The LB method is well known and on large length and timescales it leads to a hydrodynamic flow field that satisfies the Navier-Stokes equation. If the lattice fluid should be coupled to a conventional MD simulation of the coarse-grained particles, it is necessary to thermalize the fluid. While the MD particles are easily coupled via friction terms to the fluid, the correct thermalization of the lattice fluid requires to switch into mode space, which makes thermalized LB more complex and computationally expensive.
{kind=link}
{kind=link}
{kind=link}
However, the LB method is particularly well suited for the highly parallel architecture of graphics processors (GPUs). I am working on a fully thermalized GPU-LB implementation which is coupled to a MD that is running on a conventional CPU using the simulation package ESPResSo [1]. This implementation is on a single NVIDIA GTX480 or C2050 about 50 times faster than on a recent INTEL XEON E5620 quadcore, therefore replacing a full compute rack by a single desktop PC with a highend graphics card. Furthermore, due to communication overhead problems of the LB CPU code, the performance of a single NVIDIA Tesla C2050 can not achieved. Performance measurements using a AMD Opteron CPU cluster (1.9GHz) showed, that even to 96 CPU nodes are up to 12 times slower then a single GPU.
Publications
-
Bertrand Rouet-Leduc, Kipton Barros, Emmanuel Cieren, Venmugil Elango, Christoph Junghans, Turab Lookman, Jamaludin Mohd-Yusof, Robert S. Pavel, Axel Y. Rivera, Dominic Röhm, Allen L. McPherson, Timothy C. Germann.
Spatial adaptive sampling in multiscale simulation.
Computer Physics Communications (7):1857–1864, 2014.
[URL] -
D. Roehm, A. Arnold.
Lattice Boltzmann simulations on GPUs with ESPResSo.
European Physical Journal Special Topics 210(1):89–100, 2012.
[PDF] (419 KB) [DOI] -
Dominic Roehm, Kai Kratzer, Axel Arnold.
Heterogeneous and Homogeneous Crystallization of Soft Spheres in Suspension.
In High Performance Computing in Science and Engineering '13, pages 33–52. Edited by Wolfgang E. Nagel, Dietmar H. Kröner, Michael M. Resch.
Springer International Publishing, 2013. ISBN: 978-3-319-02165-2.
[DOI] -
Axel Arnold, Olaf Lenz, Stefan Kesselheim, Rudolf Weeber, Florian Fahrenberger, Dominic Röhm, Peter Košovan, Christian Holm.
ESPResSo 3.1 – Molecular Dynamics Software for Coarse-Grained Models.
In Meshfree Methods for Partial Differential Equations VI, pages 1–23. Edited by Michael Griebel, Marc Alexander Schweitzer. Part of Lecture Notes in Computational Science and Engineering, volume 89.
Springer Berlin Heidelberg, 2013.
[PDF] (380 KB) [DOI]
Curriculum vitae
Scientific education
- May 2011 - ... Doctorate studies at Institute for Computational Physics (University of Stuttgart)
- Aug. 2011 Win of the NVIDIA Best Program Award, a CUDA competition held at the 20th International Conference on Discrete Simulation of Fluid Dynamics 2011, Fargo, USA.
- May 2011 Diploma in Physics at University of Stuttgart
 Diplomarbeit (2.19 MB)
Diplomarbeit (2.19 MB)
- Oct. 2005 - May 2011 Studies of Physics at the University of Stuttgart
- June 2013 - Nov. 2013 Internship at the Los Alamos National Laboratory, USA